:format(webp)/Option_1_2_2d9677e5fd.png)
:format(webp)/Option_1_1_2a84e0cd00.png)

:format(webp)/tran_huynh_minh_nhat_905ca436dd.png)
Dược sĩ chuyên khoa Dược lý - Dược lâm sàng. Tốt nghiệp 2 trường đại học Mở và Y Dược TP. Hồ Chí Minh. Có kinh nghiệm nghiên cứu về lĩnh vực sức khỏe, đạt được nhiều giải thưởng khoa học. Hiện là Dược sĩ chuyên môn phụ trách xây dựng nội dung và triển khai dự án đào tạo - Hội đồng chuyên môn tại Nhà thuốc Long Châu.
Đa u tuyến nội tiết týp 1 (MEN1)
21/06/2023
Kích thước chữ
Mặc định
Lớn hơn
Đa u tuyến nội tiết týp 1 (Multiple Endocrine Neoplasia Type 1, MEN1) là bệnh lý di truyền liên quan đến những khối u của các tuyến nội tiết (là nơi sản xuất các hormone).
MEN 1 chủ yếu liên quan đến sự tăng sản hoặc đôi khi là u tuyến của tuyến cận giáp nguyên phát (gây cường cận giáp) và các khối u tế bào đảo tụy và/hoặc tuyến yên.
Đa u tuyến nội tiết týp 1 (Multiple Endocrine Neoplasia Type 1, MEN1) là gì?
Đa u tuyến nội tiết týp 1 (Multiple Endocrine Neoplasia Type 1, MEN1) là bệnh lý di truyền liên quan đến những khối u của các tuyến nội tiết (là nơi sản xuất các hormone). Ban đầu MEN1 còn được gọi là hội chứng Werner. Những khối u thường gặp nhất trong MEN1 bắt nguồn từ tuyến cận giáp, các tế bào đảo tuyến tụy và tuyến yên. Ngoài ra còn có những khối u nội tiết khác như u vỏ thượng thận (adrenal cortical tumors), u thần kinh nội tiết (trước đây gọi là u carcinoid) (neuroendocrine tumors), và hiếm hơn là u tủy thượng thận (pheochromocytomas) cũng như các khối u ở những phần khác của hệ tiêu hóa.
Các khối u không phải nội tiết cũng được tìm thấy trong MEN1, bao gồm:
- U sợi mạch máu (angiofibroma) vùng mặt, là khối u của mạch máu và mô sợi.
- U tế bào sản xuất collagen (collagenoma), là khối u có màu hồng nhạt trên da.
- U mỡ (lipoma).
- U cơ trơn (leiomyoma).
- U màng não (meningioma), là khối u từ mô hệ thống thần kinh; không phổ biến.
- U tế bào màng nội tủy (ependymoma), là khối u từ mô hệ thống thần kinh; không phổ biến.
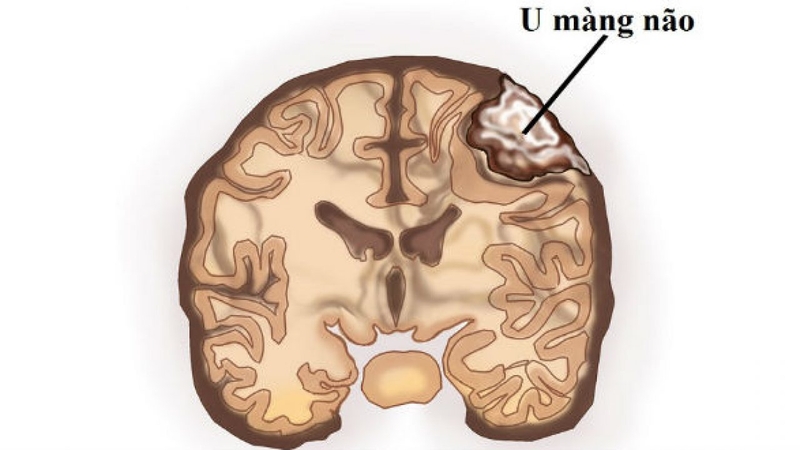 U màng não không phải nội tiết cũng được tìm thấy trong MEN1
U màng não không phải nội tiết cũng được tìm thấy trong MEN1Phần lớn các khối u ở những người mắc MEN1 đều lành tính (không ung thư). Tuy nhiên, khoảng 1 trong 3 khối u thần kinh nội tiết tuyến tụy và khối u thần kinh nội tiết trung thất là u ác tính, nghĩa là chúng có thể lan sang các cơ quan khác của cơ thể. Những khối u này có thể sản xuất lượng lớn hormone gây ra các vấn đề sức khỏe, bao gồm tăng sản xuất các hormone:
- Prolactin, việc tạo sữa của tuyến vú trở nên bất thường, mất kinh nguyệt ở nữ giới và giảm sản xuất testosterone ở nam giới.
- Hormone tăng trưởng, gây ra sự tăng trưởng quá mức của hàm và các mô mềm khác.
- hormone kích vỏ thượng thận, khiến tuyến thượng thận tăng sản xuất cortisol quá mức.
- Gastrin, gây loét dạ dày.
- Glucagon, gây ra bệnh đái tháo đường và phát ban da.
- Peptide ruột vận mạch (vasoactive intestinal peptide, VIP), được sản xuất bởi một khối u thần kinh nội tiết tuyến tụy, gây ra tiêu chảy nước dữ dội (intense watery diarrhea).
- Hormon tuyến cận giáp, được sản xuất bởi các khối u tuyến cận giáp, gây tăng canxi máu (hypercalcemia) và sỏi thận.
Nguyên nhân gây bệnh MEN1?
MEN1 là bệnh lý di truyền, có nghĩa là nguy cơ ung thư và các đặc điểm khác của MEN1 có thể được truyền từ thế hệ này sang thế hệ khác trong gia đình. Các gen liên quan đến MEN1 cũng được gọi là MEN1. Đột biến trong gen MEN1 làm tăng nguy cơ xuất hiện khối u nội tiết và các triệu chứng khác của MEN1. Hơn 90% người mang đột biến gen MEN1 sẽ biểu hiện 1 hoặc nhiều triệu chứng của MEN1. Một tỷ lệ nhỏ người không mang đột biến gen MEN1 được phát hiện đang mang đột biến trong tế bào mầm (đột biến trong trứng hoặc tế bào tinh trùng và được di truyền trong ADN của mọi tế bào), ảnh hưởng đến các protein ức chế kinase phụ thuộc cyclin (cyclin-dependent kinase inhibitors, CDKIs). Đây là các protein có vai trò điều chỉnh sự phát triển và phân chia của tế bào. Các nghiên cứu đang được tiếp tục để tìm hiểu thêm về MEN1.
MEN1 di truyền như thế nào?
Thông thường, mỗi tế bào có 2 bản sao của mỗi gen: 1 bản sao được thừa hưởng từ mẹ và 1 bản sao được thừa hưởng từ cha. MEN1 là bệnh di truyền trội trên nhiễm sắc thể thường, có nghĩa là đột biến chỉ cần xảy ra trong 1 bản sao của gen thì cũng sẽ biểu hiện bệnh. Nếu 1 bản sao của gen có đột biến được di truyền, gen đó trở thành gen trội và có thể biểu hiện các đặc điểm của bệnh MEN1. Người cha hoặc mẹ mắc MEN1 có thể truyền cho con mình 1 bản sao của gen bình thường hoặc 1 bản sao của gen mang đột biến. Tuy nhiên, nếu cha mẹ xét nghiệm di truyền cho kết quả âm tính, nguy cơ đối với anh chị em ruột giảm đáng kể nhưng rủi ro của họ vẫn có thể cao hơn mức trung bình.
Những bậc cha mẹ tương lai đang mang gen đột biến khi quyết định có con sẽ làm tăng nguy cơ của hội chứng ung thư di truyền này. Để biết thêm thông tin, hãy trao đổi với chuyên gia hỗ trợ sinh sản tại phòng khám.
MEN1 có phổ biến?
Ước tính có khoảng 1 trên 30.000 người mắc MEN1. Khoảng 10% những người mắc MEN1 không có tiền sử gia đình mắc bệnh này; họ mang những đột biến gen MEN1 mới.
Làm thế nào để chẩn đoán MEN1?
Nghĩ đến MEN1 khi một người mắc ít nhất 2 trong số những khối u phổ biến nhất được liệt kê dưới đây:
- Khối u tuyến cận giáp;
- Khối u thần kinh nội tiết tuyến tụy;
- Khối u tuyến yên.
Nếu một người có tiền sử gia đình mắc MEN1, cần nghi ngờ bệnh MEN1 nếu người đó mắc khối u tuyến cận giáp, tuyến tụy hoặc khối u tuyến yên. Có thể xét nghiệm di truyền phát hiện đột biến gen MEN1 cho những người nghi ngờ mắc MEN1. Có thể phát hiện đột biến gen MEN1 ở khoảng 80% – 90% các gia đình được chẩn đoán mắc MEN1. Đột biến gen MEN1 có ở 65% những người mắc từ 2 khối u trở lên liên quan đến MEN1 nhưng không có tiền sử gia đình mắc MEN1.
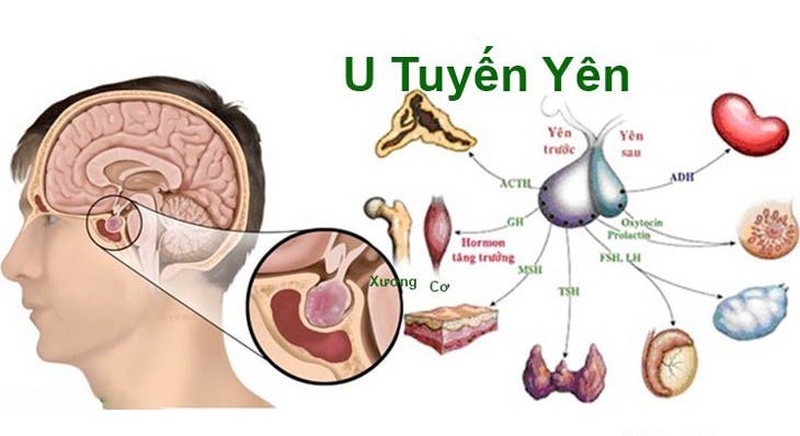 Khối u tuyến yên là khối u phổ biến
Khối u tuyến yên là khối u phổ biếnƯớc tính nguy cơ ung thư từ bệnh MEN1?
Khoảng 1/3 khối u thần kinh nội tiết tuyến tụy là ác tính. Ung thư thường di căn nhiều nhất đến gan. Một tỷ lệ nhỏ các khối u thần kinh nội tiết trung thất là ác tính và lan đến các hạch bạch huyết tại chỗ (gần đó) hoặc di căn đến gan, phổi hoặc các vị trí khác.
Những lựa chọn sàng lọc cần cho bệnh nhân mắc MEN1?
Những sàng lọc cho bệnh nhân mắc MEN1 hoặc nghi ngờ đang mắc MEN1 bao gồm:
Xét nghiệm di truyền
- Xét nghiệm di truyền được xem xét cho trẻ em hoặc thanh niên có tiền sử gia đình mắc MEN1. Xác định đột biến gen MEN1 để quyết định thực hiện các xét nghiệm sàng lọc được mô tả dưới đây. Trẻ có tiền sử gia đình mắc MEN1 và xét nghiệm di truyền cho thấy trẻ không mang đột biến MEN1 (kỳ vọng ở 50% trẻ sinh ra bởi người mắc MEN1) có thể không cần các xét nghiệm sàng lọc được mô tả dưới đây.
Xét nghiệm chẩn đoán
- Xét nghiệm máu thường xuyên mỗi 1 – 3 năm, định lượng prolactin, yếu tố tăng trưởng giống insulin 1 (insulin-like growth factor 1, IGF-1), đường huyết lúc đói, insulin và proinsulin, bắt đầu khi 5 – 10 tuổi.
- Xét nghiệm nồng độ canxi dạng ion (ionized calcium level) hoặc nồng độ canxi hiệu chỉnh theo albumin máu (albumin-corrected calcium level) hàng năm, bắt đầu khi 8 tuổi
- Xét nghiệm máu thường xuyên để định lượng gastrin lúc đói, polypeptide tụy (pancreatic polypeptide, PP) lúc đói và sau ăn, glucagon và peptide ruột vận mạch (vasoactive intestinal peptide, VIP) lúc đói, bắt đầu từ năm 20 tuổi.
- Chụp cộng hưởng từ (MRI) não, sau mỗi 3 – 5 năm, bắt đầu khi 5 – 10 tuổi, hoặc bất cứ lúc nào kết quả xét nghiệm máu cho kết quả bất thường về nồng độ prolactin hoặc yếu tố tăng trưởng giống insulin (insulin-like growth factor 1, IGF-1).
- Chụp MRI hoặc chụp cắt lớp vi tính (CT) vùng ngực và bụng, sau mỗi 2 – 4 năm, bắt đầu khi 20 tuổi hoặc khi bất thường về nồng độ gastrin, PP hoặc VIP máu.
Các khuyến cáo sàng lọc có thể thay đổi theo thời gian, khi các công nghệ mới được phát triển và hiểu biết nhiều hơn về MEN1. Bạn cần trao đổi với Bác sĩ để chọn lựa xét nghiệm sàng lọc thích hợp.
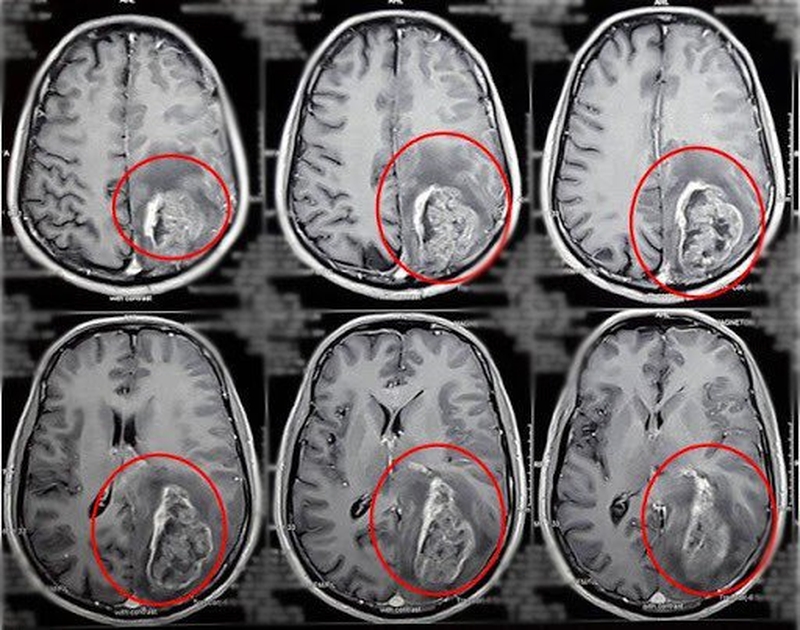 Chụp cộng hưởng từ (MRI) não, sau mỗi 3 – 5 năm, bắt đầu khi 5 – 10 tuổi
Chụp cộng hưởng từ (MRI) não, sau mỗi 3 – 5 năm, bắt đầu khi 5 – 10 tuổiPhương pháp điều trị những khối u nội tiết?
Hầu hết những khối u này được điều trị bằng phẫu thuật hoặc những thuốc có tác dụng ngăn sự phát triển hoặc những tác động của khối u. Hầu hết khối u tuyến cận giáp là lành tính, vì vậy có thể phẫu thuật cắt bỏ khi nồng độ canxi hiệu chỉnh theo albumin lớn hơn 12 mg/dl, khi có dấu hiệu tiêu xương đáng kể, dấu hiệu tổn thương thận hoặc có sỏi. Việc loại bỏ các khối u thần kinh nội tiết tuyến tụy là một thách thức trong điều trị. Tuyến tụy không chỉ có vai trò trong tiêu hóa bình thường mà còn giúp điều chỉnh nồng độ glucose máu thông qua sản xuất insulin.
Cắt bỏ tuyến tụy sẽ gây ra bệnh đái tháo đường (đây là bệnh lý gây những biến chứng nghiêm trọng) và cần phải bổ sung enzyme tuyến tụy để đảm bảo chức năng tiêu hóa. Các bác sĩ phải cân bằng lợi ích của việc cắt bỏ tụy ở người mắc MEN1, chẳng hạn như ngăn ngừa sự phát triển của ung thư, chống lại nguy cơ mắc bệnh đái tháo đường. Bệnh nhân có khối u thần kinh nội tiết tuyến tụy đã di căn đến gan có thể được điều trị bằng chất tương tự somatostatin (somatostatin analogue) hoặc một loại thuốc điều chỉnh tín hiệu trong tế bào đảo tụy, everolimus. Các khối u thần kinh nội tiết khác thường được loại bỏ bằng phẫu thuật và có thể sử dụng những phương pháp điều trị khác.
Các khối u tuyến yên sản xuất hormone prolactin được điều trị với thuốc đồng vận dopamine (dopamine agonist) – là thuốc bắt chước hoạt động của dopamine (dopamine là chất tự nhiên được sản xuất trong não). Các khối u sản xuất hormone tăng trưởng, hormone kích vỏ thượng thận hoặc những khối u không hoạt động thường được điều trị bằng phẫu thuật. Đối với khối u tăng tiết quá nhiều hormone tăng trưởng, nếu không chữa khỏi bằng phẫu thuật, có thể điều trị thành công bằng 2 liệu pháp hormone là sử dụng các hợp chất tương tự somatostatin (somatostatin analogue) và chất đối kháng hormone tăng trưởng (growth hormone antagonist).
Thủy Phan
Nguồn tham khảo: yhoccongdong.com
Có thể bạn quan tâm
Thông tin và sản phẩm gợi ý trong bài viết chỉ mang tính chất tham khảo, vui lòng liên hệ với Bác sĩ, Dược sĩ hoặc chuyên viên y tế để được tư vấn cụ thể. Xem thêm
Các bài viết liên quan
WHO cảnh báo số ca ung thư có thể tăng gần gấp đôi vào năm 2050
6 vật dụng tưởng vô hại nhưng tiềm ẩn nguy cơ gây ung thư
3 dấu hiệu ung thư tuyến tụy nhiều người Việt chỉ nghĩ là rối loạn tiêu hóa bình thường
Uống thuốc phóng xạ thì bao lâu mới được quan hệ?
Phát hiện mới về hệ miễn dịch có thể giúp chống lại tế bào ung thư
Thực đơn cho người ung thư phổi giai đoạn cuối giúp cải thiện thể trạng
Các triệu chứng ung thư dạ dày giai đoạn đầu thường gặp
Nguyên nhân gây ung thư phổi và các yếu tố nguy cơ không ngờ tới
Ung thư phổi di căn xương và những điều cần biết
Ung thư biểu mô tuyến kém biệt hóa là gì? Thông tin cần biết