:format(webp)/Option_1_2_2d9677e5fd.png)
:format(webp)/Option_1_1_2a84e0cd00.png)

:format(webp)/ds_my_huyen_780f9bef46.png)
Dược sĩ Đại học có nhiều năm kinh nghiệm trong việc tư vấn Dược phẩm và hỗ trợ giải đáp thắc mắc về Bệnh học. Hiện đang là giảng viên cho Dược sĩ tại Nhà thuốc Long Châu.
Phenylketone niệu (PKU): Nguyên nhân, triệu chứng và điều trị
Chí Doanh
16/07/2024
Kích thước chữ
Mặc định
Lớn hơn
PKU là một bệnh di truyền bẩm sinh về rối loạn chuyển hóa axit amin phổ biến. Tỷ lệ mắc bệnh là như nhau ở cả nam và nữ. Vậy PKU là gì? PKU có chữa được không? Hãy cùng Nhà thuốc Long Châu tìm hiểu về căn bệnh này qua bài viết dưới đây.
Phenylalanine là axit amin thiết yếu để sản xuất protein. Nếu phenylalanine tích lũy vượt mức cho phép sẽ ảnh hưởng đến sự phát triển và chức năng của não. Trong y học, có một bệnh gọi là phenylketone niệu làm tăng quá mức phenylalanine và gây ra hậu quả nặng nề nếu không điều trị.
Phenylketone niệu (PKU) là gì?
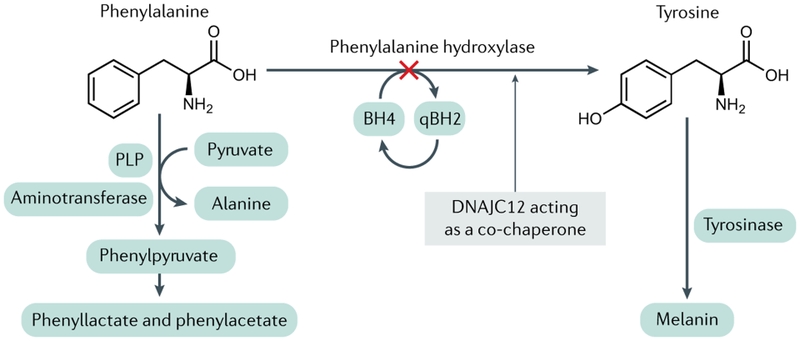
Phenylketon niệu (PKU) là bệnh rối loạn chuyển hóa phenylalanine do thiếu hụt phenylalanine hydroxylase (PAH). Phenylalanine hydroxylase xúc tác quá trình hydroxyl hóa L-phenylalanine (Phe) thành L-tyrosine (Tyr), một phản ứng xảy ra chủ yếu ở gan nhưng cũng xảy ra ở ống lượn gần ở thận. Ở những bệnh nhân có kiểu gen PAH lặn trên nhiễm sắc thể thường, hoạt động của enzyme PAH hoàn toàn thiếu hoặc bị giảm nghiêm trọng, gây ra sự tích tụ Phe trong cơ thể, khiến nồng độ Phe tăng cao gây ra rối loạn chức năng não và nồng độ tyrosine cũng giảm đi so với nồng độ ở những người có đủ PAH.
Ở những bệnh nhân mắc PKU không được điều trị, nồng độ Phe trong máu tăng lên rõ rệt, dẫn đến hình thành các thể phenylketone được bài tiết qua nước tiểu. Về mặt lâm sàng, những bệnh nhân không được điều trị sẽ bị thiểu năng trí tuệ nghiêm trọng, động kinh và các vấn đề về hành vi, tâm thần và vận động, cũng như da, mắt và tóc có sắc tố nhẹ, bệnh chàm.
Nguyên nhân gây bệnh PKU
Phenylalanine là một trong những axit amin thiết yếu cho cơ thể con người. Lượng tiêu thụ hàng ngày của người bình thường là khoảng 200 đến 500 mg Phe, nó dùng để tổng hợp protein và chuyển hóa thành tyrosine thông qua phenylalanine hydroxylase trong tế bào gan. Tyr có mặt trong một số quá trình trao đổi chất, bao gồm sản xuất các chất dẫn truyền thần kinh dopamine, adrenaline và norepinephrine, chuyển đổi thành thyroxine trong tuyến giáp và thành melanin trong tế bào hắc tố, đồng thời dị hóa hoàn toàn thành acetoacetate (ketone) và fumarate,...
Trong quá trình chuyển hóa phenylalanine thành tyrosine, ngoài PAH, tetrahydrobiopterin (BH4) cũng phải tham gia với tư cách là coenzym. Đột biến gen PAH nằm trên nhiễm sắc thể 12 là nguyên nhân gây ra khiếm khuyết trong hoạt động của các enzyme liên quan, dẫn đến sự tích tụ phenylalanine bất thường.
Biểu hiện lâm sàng của PKU
Các biểu hiện lâm sàng chỉ xảy ra khi bệnh nhân PKU không điều trị hoặc không thể kiểm soát tốt khi đã điều trị.
Chậm tăng trưởng và phát triển
Ngoài chậm phát triển thể chất còn biểu hiện chủ yếu ở chậm phát triển trí tuệ. Nó biểu hiện ở mức IQ thấp hơn so với trẻ bình thường cùng tuổi và có thể xuất hiện từ 4 đến 9 tháng sau khi sinh. Trong trường hợp nghiêm trọng, chỉ số IQ thấp hơn 50 và rối loạn phát triển ngôn ngữ đặc biệt rõ ràng, những triệu chứng này cho thấy rối loạn phát triển não bộ.
Biểu hiện thần kinh tâm thần
Có những dị tật tiểu não do teo não và co giật tái phát nhưng chúng giảm dần theo tuổi tác. Tăng trương lực cơ và tăng phản xạ. Thường có hưng phấn, bồn chồn, hiếu động thái quá và có hành vi bất thường.
Biểu hiện về da và tóc
Da thường bị khô và dễ bị chàm, tổn thương. Do ức chế tyrosinase, quá trình tổng hợp melanin bị giảm nên màu tóc của trẻ có màu nhạt và nâu.
Biểu hiện khác
Do thiếu phenylalanine hydroxylase, phenylalanine tăng dần tạo ra axit phenyllactic và axit phenylacetic theo con đường khác và được bài tiết qua mồ hôi và nước tiểu khiến nước tiểu và mồ hôi có mùi.
Nếu điều trị không hiệu quả trong thời gian dài, người lớn mắc PKU có thể phát triển các vấn đề lâm sàng, bao gồm co cứng chi dưới và mất điều hòa tiểu não, run, bệnh não và bất thường về thị giác.

Các phương pháp chẩn đoán PKU
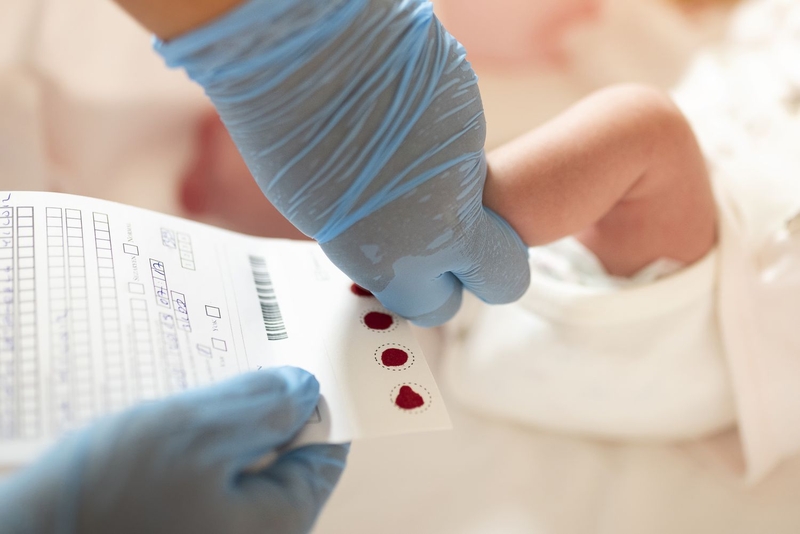
Sàng lọc sơ sinh
Ba ngày sau sinh, trẻ sơ sinh được thu thập máu từ gót chân và gửi đến trung tâm sàng lọc. Xét nghiệm ức chế tăng trưởng vi khuẩn Guthrie được sử dụng để đo bán định lượng. Nguyên tắc là phenylalanine có thể thúc đẩy sự phát triển của vi khuẩn bị ức chế, Bacillus subtilis phát triển trở lại và hàm lượng phenylin trong máu được đo trong phạm vi của vòng tròn tăng trưởng.
Phân tích DNA
Trong những năm gần đây, phân tích DNA đã được sử dụng rộng rãi trong chẩn đoán PKU và chẩn đoán trước sinh để phát hiện dị hợp tử. Tuy nhiên, do tính đa hình di truyền nên kết quả phân tích phải thận trọng.
PKU có điều trị được không?
PKU không được điều trị chỉ được thấy ở những người được sinh ra trước khi triển khai sàng lọc sơ sinh hoặc ở một quốc gia mà việc sàng lọc sơ sinh chưa được thực hiện cho mọi trẻ sơ sinh và do đó không được điều trị từ khi sinh ra. Nếu bắt đầu ngay sau khi sinh, điều trị bằng chế độ ăn kiêng có thể ngăn ngừa những di chứng của bệnh PKU.
Việc điều trị bằng chế độ ăn uống bao gồm ba khía cạnh:
- Hạn chế lượng Phe tự nhiên: Vì phenylalanine là một axit amin thiết yếu để tổng hợp protein nên nếu thiếu hụt hoàn toàn cũng có thể gây tổn thương cho hệ thần kinh, vì vậy, trẻ nhỏ có thể được cho ăn thức ăn có hàm lượng phenylalanine thấp. Trong quá trình điều trị bằng chế độ ăn hạn chế lượng Phe, cần phải theo dõi chặt chẽ sự tăng trưởng, phát triển và tình trạng dinh dưỡng của trẻ cũng như nồng độ phenylalanine trong máu và các tác dụng phụ. Tác dụng phụ chính là sự thiếu hụt dinh dưỡng khác, có thể bao gồm tiêu chảy, thiếu máu, hạ đường huyết và phát ban giống thiếu niacin.
- Bổ sung hỗn hợp axit amin không chứa Phe: Việc bổ sung tyrosine trong chế độ ăn uống có thể trắc phục tình trạng mất sắc tố tóc, nhưng không có tác dụng đối với sự phát triển trí tuệ.
- Tiêu thụ các sản phẩm thực phẩm ít protein: Tăng tiêu thụ các thực phẩm carbohydrate và chất béo tốt để cung cấp đủ năng lượng hoạt động và giúp bệnh nhân có chế độ ăn uống giống với thói quen ăn uống bình thường ở một mức độ nào đó.
Tóm lại, phenylketone niệu (PKU) là một bệnh di truyền lặn trên nhiễm sắc thể thường do thiếu hụt phenylalanine hydroxylase ở gan, dẫn đến không có khả năng chuyển hóa phenylalanine. Các đặc điểm lâm sàng là chậm phát triển trí tuệ, các triệu chứng tâm thần kinh, chàm,... Những biểu hiện lâm sàng này có thể tránh được bằng cách chẩn đoán và điều trị sớm.
Xem thêm:
Thông tin và sản phẩm gợi ý trong bài viết chỉ mang tính chất tham khảo, vui lòng liên hệ với Bác sĩ, Dược sĩ hoặc chuyên viên y tế để được tư vấn cụ thể. Xem thêm
Các bài viết liên quan
Tiểu không hết do bệnh gì? Nguyên nhân và cách chữa hiệu quả
Giãn bàng quang là gì? Nguyên nhân, dấu hiệu và cách điều trị
Bộ Y tế đề xuất sàng lọc 90 bệnh bẩm sinh, mở rộng can thiệp sớm
Phương pháp ngủ trưa giúp thận thải độc và phục hồi
5 cách khắc phục đi tiểu nhiều lần giúp cải thiện sinh hoạt hằng ngày
Kết quả sàng lọc trước sinh nguy cơ thấp có đáng lo?
Nguyên nhân gây sa niệu đạo nữ và các phương pháp điều trị hiệu quả
Mắc tiểu liên tục nhưng tiểu ít ở nữ: Nguyên nhân và cách điều trị hiệu quả
Đi tiểu liên tục cảnh báo bệnh gì? Có nguy hiểm không?
Sàng lọc trước sinh gồm những xét nghiệm nào để an toàn thai kỳ?