:format(webp)/Option_1_2_2d9677e5fd.png)
:format(webp)/Option_1_1_2a84e0cd00.png)

:format(webp)/Duoc_si_nguyen_vu_kieu_ngan_48fc1f2f55.png)
Tốt nghiệp Đại học Y Dược TP. Hồ Chí Minh. Có nhiều năm trong lĩnh vực dược phẩm. Hiện đang là giảng viên cho Dược sĩ tại Nhà thuốc Long Châu.
Hội chứng Hurler: Nguyên nhân, dấu hiệu nhận biết và cách điều trị hiệu quả
Thanh Hương
20/01/2026
Kích thước chữ
Mặc định
Lớn hơn
Trong số các bệnh rối loạn chuyển hóa hiếm gặp, hội chứng Hurler là một trong những căn bệnh nguy hiểm nhất, ảnh hưởng nghiêm trọng đến sức khỏe và sự phát triển của trẻ em. Bài viết sẽ cung cấp những thông tin chi tiết để các bậc cha mẹ hiểu rõ hơn về hội chứng này.
Hội chứng Hurler là một bệnh lý di truyền hiếm gặp nhưng nghiêm trọng, ảnh hưởng trực tiếp đến sự phát triển thể chất và tinh thần của trẻ. Bệnh gây ra do sự thiếu hụt enzym cần thiết để phân giải các phân tử đường phức tạp, dẫn đến tích tụ chất độc hại trong cơ thể và tổn thương nhiều cơ quan. Bài viết này sẽ cung cấp thông tin chi tiết về nguyên nhân, các dấu hiệu nhận biết cũng như phương pháp điều trị hội chứng Hurler, giúp cải thiện chất lượng sống cho trẻ.
Hội chứng Hurler là gì?
Hội chứng Hurler là một dạng rối loạn dự trữ lysosome xảy ra khi cơ thể bị thiếu hụt enzyme α-L-iduronidase. Đây là loại enzyme có vai trò quan trọng trong việc phân giải một số chất đường phức tạp gọi là mucopolysaccharid. Khi enzyme này không hoạt động bình thường, dẫn đến tích tụ glycosaminoglycans - chủ yếu dermatan sulfate và heparan sulfate, gây ảnh hưởng đến nhiều cơ quan và hệ thống trong cơ thể.
Về đặc điểm di truyền, hội chứng Hurler được xếp vào nhóm bệnh di truyền lặn trên nhiễm sắc thể thường. nếu cả bố và mẹ đều là người mang đột biến, mỗi lần mang thai có 25% nguy cơ con mắc bệnh, 50% là mang gen, 25% không mang. Chính sự bất thường về di truyền này khiến mucopolysaccharid tích tụ lâu dài, làm thay đổi cấu trúc và chức năng của các mô, cơ quan.

Đặc biệt, hội chứng Hurler được coi là thể nặng nhất trong nhóm bệnh MPS (rối loạn di truyền hiếm gặp Mucopolysaccharidosis type I). Bệnh thường tiến triển nhanh chóng, gây biến chứng nặng nề trên nhiều hệ cơ quan như xương khớp, tim mạch, hô hấp và thần kinh. Nếu không được phát hiện và điều trị sớm, bệnh có thể rút ngắn đáng kể tuổi thọ và ảnh hưởng nghiêm trọng đến chất lượng sống của trẻ.
Nguyên nhân và cơ chế bệnh sinh hội chứng Hurler
Căn bệnh này xuất phát từ một đột biến xảy ra trên gen IDUA, nằm trên nhiễm sắc thể số 4. Khi gen IDUA bị đột biến, cơ thể không thể sản xuất đủ hoặc không sản xuất được enzyme α-L-iduronidase - một loại enzyme lysosomal. Lysosome là một "nhà máy tái chế" bên trong tế bào, nơi các chất thải phức tạp được phân hủy. Enzyme α-L-iduronidase có nhiệm vụ phân giải hai loại mucopolysaccharide (còn gọi là glycosaminoglycans - GAGs) gồm heparan sulfate và dermatan sulfate.
Khi enzyme này bị thiếu hụt, hai chất trên không thể được phân giải và bắt đầu tích tụ bên trong lysosome của tế bào. Sự tích tụ này giống như một bãi rác khổng lồ bên trong tế bào, dần dần làm hỏng và phá hủy tế bào. Do heparan sulfate và dermatan sulfate có mặt ở hầu hết các mô và cơ quan trong cơ thể, sự tích tụ này sẽ gây ra tổn thương đa cơ quan, ảnh hưởng nặng nề đến sự phát triển thể chất và tinh thần của trẻ.

Dấu hiệu thường gặp ở trẻ mắc hội chứng Hurler
Triệu chứng bệnh thường bắt đầu trong vài tháng đầu và tiến triển. Một số đặc điểm (ví dụ chậm phát triển thần kinh) biểu hiện sớm ở thể nặng. Dưới đây là những dấu hiệu thường gặp ở trẻ mắc hội chứng này mà cha mẹ cần đặc biệt lưu ý:
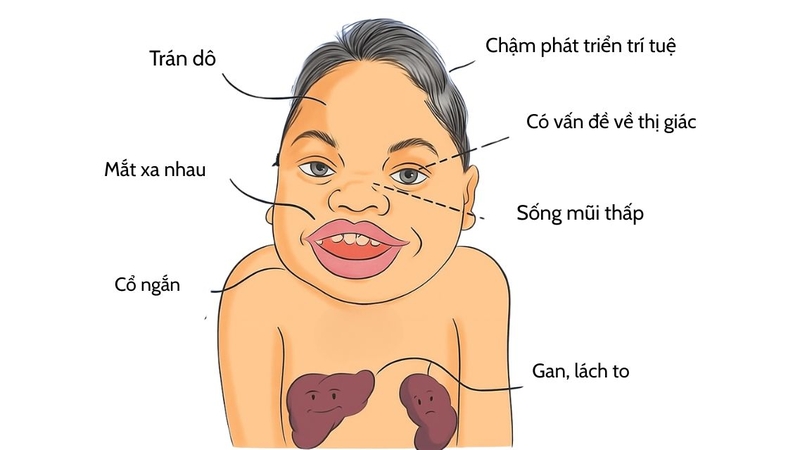
Đặc điểm khuôn mặt thô, trán dô, mũi tẹt
Trẻ mắc hội chứng Hurler thường có gương mặt đặc trưng với sống mũi thấp, môi dày, lưỡi to và trán dô. Những thay đổi này xuất hiện dần theo thời gian, giúp bác sĩ có thể gợi ý chẩn đoán sớm.
Tầm vóc thấp, chậm phát triển thể chất
Hầu hết trẻ bị Hurler có chiều cao thấp hơn so với bạn cùng lứa tuổi. Sự chậm phát triển thể chất thể hiện rõ khi trẻ lớn dần, ảnh hưởng đến khả năng vận động và sinh hoạt.
Biến dạng xương do hội chứng Hurler
Tình trạng này xảy ra do sự tích tụ mucopolysaccharid trong mô liên kết và xương. Trẻ có thể bị biến dạng cột sống (gù, vẹo), lồng ngực bất thường và khớp cứng, hạn chế vận động.
Các vấn đề thị giác và thính giác
Trẻ mắc hội chứng này có giác mạc đục, dần làm giảm thị lực. Kèm theo đó là suy giảm thính lực do viêm tai giữa tái phát hoặc tổn thương thần kinh thính giác. Hai yếu tố này ảnh hưởng lớn đến việc học tập và giao tiếp của trẻ.
Rối loạn tim mạch
Bệnh nhân Hurler thường gặp các vấn đề tim mạch nghiêm trọng như phì đại tim, dày cơ tim hoặc hở van tim. Những biến chứng này làm tăng nguy cơ suy tim và tử vong sớm.
Chậm phát triển trí tuệ, giảm khả năng học tập
Ngoài ảnh hưởng thể chất, trẻ còn gặp khó khăn về nhận thức. Khả năng ngôn ngữ, tư duy và học tập giảm dần theo thời gian, khiến trẻ dễ bị tụt hậu so với bạn bè đồng trang lứa.
Biến chứng của hội chứng Hurler nếu không điều trị
Tiên lượng của hội chứng Hurler phụ thuộc nhiều vào thời điểm phát hiện và can thiệp điều trị. Trẻ mắc Hurler thường bị hạn chế vận động, khó khăn trong học tập và giao tiếp do chậm phát triển trí tuệ, kèm theo nhiều vấn đề về thị giác, thính giác. Điều này khiến cuộc sống sinh hoạt hằng ngày của trẻ và gia đình gặp nhiều thách thức.
Khối mucopolysaccharid tích tụ lâu ngày gây tổn thương hàng loạt cơ quan. Tim có thể bị phì đại, hở van, dễ dẫn đến suy tim. Gan và lách to bất thường, hệ hô hấp bị ảnh hưởng khiến trẻ dễ viêm phổi, khó thở, ngủ ngáy. Do bệnh tiến triển nhanh và gây biến chứng toàn thân, đa số trẻ không được điều trị kịp thời có tuổi thọ ngắn, thường chỉ sống dưới 10 - 12 tuổi.
Phương pháp điều trị hội chứng Hurler
Hiện nay, hội chứng Hurler chưa có phương pháp chữa khỏi hoàn toàn, nhưng nhiều biện pháp y học đã và đang được áp dụng nhằm kéo dài tuổi thọ, cải thiện chất lượng sống cho bệnh nhân.
Ghép tủy xương
Ghép tủy xương là phương pháp đem lại lợi ích lớn, nhất là khi thực hiện sớm (thường trong năm đầu đời), giúp cung cấp nguồn tế bào sản xuất α-L-iduronidase và có thể ổn định hoặc giảm tiến triển tổn thương thần kinh. Hiệu quả về thần kinh giảm nếu ghép thực hiện muộn và ghép tủy cũng tiềm ẩn rủi ro và đòi hỏi cơ sở y tế chuyên sâu.
Liệu pháp thay thế enzyme
Liệu pháp thay thế enzyme (ERT) là phương pháp truyền enzyme α-L-iduronidase tái tổ hợp cho bệnh nhân. Liệu pháp này giúp giảm sự tích tụ glycosaminoglycans ở nhiều cơ quan ngoại biên và cải thiện các triệu chứng hệ thống. Nhưng enzyme này không qua được hàng rào máu - não ở liều thông thường, nên không kiểm soát hiệu quả bệnh lý thần kinh trung ương
Điều trị hỗ trợ
Điều trị hỗ trợ đóng vai trò quan trọng trong việc nâng cao chất lượng sống cho trẻ mắc hội chứng Hurler. Các phương pháp điều trị hỗ trợ bao gồm: Phẫu thuật chỉnh hình để cải thiện biến dạng xương, điều trị các bệnh lý tim mạch, hỗ trợ hô hấp cho trẻ bị tắc nghẽn đường thở, cũng như can thiệp về mắt và tai để hạn chế suy giảm thị lực và thính lực.
Cách phòng ngừa hội chứng Hurler
Hiện nay, không có biện pháp phòng bệnh tuyệt đối đối với hội chứng Hurler. Tuy nhiên, y học đã phát triển nhiều phương pháp giúp phát hiện sớm nguy cơ, từ đó hỗ trợ cha mẹ trong việc đưa ra quyết định sinh sản an toàn hơn. Một trong những cách quan trọng nhất là tư vấn di truyền cho các gia đình có người mắc bệnh. Khi có tiền sử trong gia đình từng xuất hiện hội chứng Hurler hoặc các rối loạn mucopolysaccharidosis khác, các cặp vợ chồng nên tham khảo ý kiến chuyên gia di truyền để hiểu rõ khả năng truyền bệnh cho con và có các lựa chọn y tế phù hợp.

Các cặp vợ chồng thuộc nhóm nguy cơ cao nên thực hiện sàng lọc di truyền hoặc sàng lọc trước sinh. Các kỹ thuật như chọc ối, sinh thiết gai nhau hoặc xét nghiệm di truyền tiền làm tổ (PGD) có thể phát hiện sớm đột biến gen gây bệnh, từ đó cho phép bác sĩ và gia đình có kế hoạch chăm sóc hoặc can thiệp kịp thời.
Việc phát hiện sớm và can thiệp kịp thời là yếu tố then chốt để cải thiện chất lượng cuộc sống cho trẻ mắc hội chứng Hurler. Với sự tiến bộ của khoa học, các phương pháp điều trị như liệu pháp enzyme thay thế hay cấy ghép tủy xương đã mang lại hy vọng cho nhiều gia đình.
Xem thêm: Hội chứng Gianotti Crosti: Bệnh lý về da hiếm gặp ở trẻ em cha mẹ cần biết
Thông tin và sản phẩm gợi ý trong bài viết chỉ mang tính chất tham khảo, vui lòng liên hệ với Bác sĩ, Dược sĩ hoặc chuyên viên y tế để được tư vấn cụ thể. Xem thêm
Các bài viết liên quan
Đặt cây kim tiền để trong phòng ngủ có tốt không? Lợi ích của việc đặt cây trong phòng ngủ
Uống dầu dừa trước khi đi ngủ có tốt không? Hướng dẫn sử dụng dầu dừa trước khi đi ngủ
Cả nhà dễ bệnh tật, ung thư chỉ vì 6 thói quen trong bếp
Hình ảnh tiêu xương ổ răng nhận biết mức độ tổn thương
Thể dục thủy sinh là gì? Những lợi ích cho sức khỏe bạn nên biết
Tầm vóc thấp bé bẩm sinh (loạn sản sụn): Nguyên nhân, dấu hiệu và cách sàng lọc sớm
Cháo sò lông: Món ăn bổ dưỡng và cách chế biến tại nhà
Cháo cá chép: Món ăn bổ dưỡng và cách nấu không bị tanh
Biểu hiện hội chứng Patau và phương pháp phòng ngừa cần biết
Hội chứng Mayer Rokitansky Kuster Hauser: Nguyên nhân và điều trị