:format(webp)/Option_1_2_2d9677e5fd.png)
:format(webp)/Option_1_1_2a84e0cd00.png)

:format(webp)/Duoc_si_nguyen_vu_kieu_ngan_48fc1f2f55.png)
Tốt nghiệp Đại học Y Dược TP. Hồ Chí Minh. Có nhiều năm trong lĩnh vực dược phẩm. Hiện đang là giảng viên cho Dược sĩ tại Nhà thuốc Long Châu.
Hạ betalipoprotein máu gia đình: Nguy cơ tiềm ẩn và những điều cần biết
Thanh Hương
25/06/2025
Kích thước chữ
Mặc định
Lớn hơn
Hạ betalipoprotein máu gia đình là rối loạn di truyền hiếm gặp. Bệnh gây ra hàng loạt triệu chứng từ rối loạn tiêu hóa đến tổn thương thần kinh, đe dọa nghiêm trọng sức khỏe nếu không chẩn đoán sớm. Bài viết sẽ cung cấp thông tin về nguyên nhân, dấu hiệu nhận biết và cách quản lý, điều trị bệnh hiệu quả.
Hạ betalipoprotein máu gia đình là một rối loạn di truyền hiếm gặp ảnh hưởng đến sự chuyển hóa lipid trong cơ thể. Mặc dù bệnh này có thể không biểu hiện rõ rệt trong giai đoạn đầu, nhưng những ảnh hưởng lâu dài đến sức khỏe tim mạch và hệ thống lipid của cơ thể lại vô cùng nghiêm trọng. Trong bài viết này, chúng ta sẽ cùng tìm hiểu về nguyên nhân, tác động, triệu chứng và cách điều trị tình trạng hạ betalipoprotein máu gia đình.
Tổng quan về hạ betalipoprotein máu gia đình
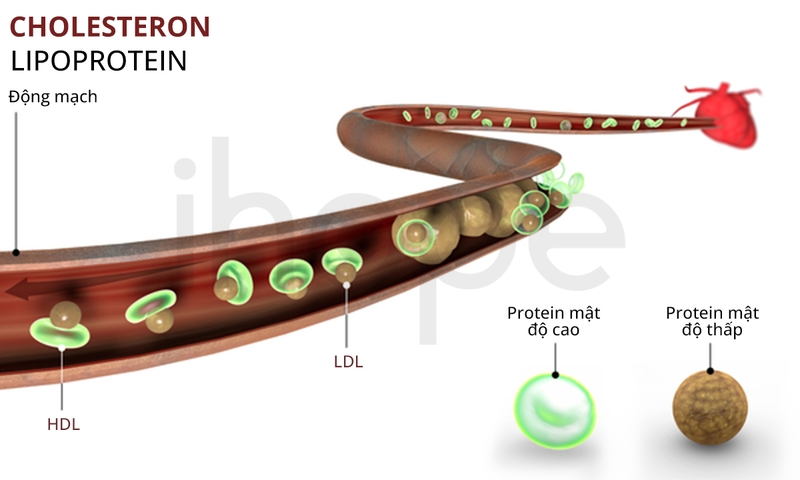
Hạ betalipoprotein máu gia đình (Familial Hypobetalipoproteinemia - FHBL) là rối loạn di truyền hiếm gặp, gây ra tình trạng thiếu hụt betalipoprotein. Khi betalipoprotein thiếu hụt, quá trình hấp thu chất béo và vitamin tan trong dầu (A, D, E, K) bị gián đoạn, dẫn đến tích tụ lipid trong tế bào gan và gây ra hàng loạt rối loạn chuyển hóa. Với tỷ lệ mắc khoảng 1/1.000.000, FHBL phổ biến hơn ở người gốc Địa Trung Hải hoặc Bắc Phi. Trên lâm sàng, bệnh nhân xét nghiệm máu thấy LDL-C <50 mg/dL và triglyceride <30 mg/dL, kèm giảm apolipoprotein B.
Tiên lượng bệnh phụ thuộc vào việc tuân thủ điều trị. Khoảng 70% bệnh nhân ổn định nhờ bổ sung vitamin liều cao, chế độ ăn giàu chất béo chuỗi trung bình (MCT) và theo dõi định kỳ. Tuy nhiên, 30% trường hợp tiến triển nặng với biến chứng như xơ gan, thoái hóa võng mạc gây mù lòa, hoặc rối loạn vận động do tổn thương tiểu não.
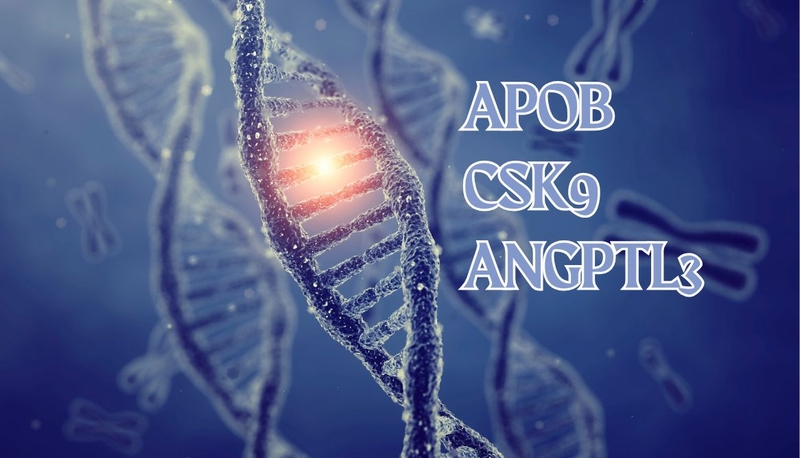
Nguyên nhân hạ betalipoprotein máu gia đình
FHBL chủ yếu do đột biến ở gen APOB. Đột biến ở các gen khác như PCSK9 và ANGPTL3 có thể liên quan đến các rối loạn lipid máu khác. Đột biến gen APOB làm giảm tổng hợp apolipoprotein B – thành phần cấu trúc thiết yếu của LDL và VLDL, khiến cơ thể không vận chuyển được cholesterol và triglyceride từ gan vào máu.
Trong khi đó, đột biến mất chức năng ở gen PCSK9 làm giảm sự phân hủy thụ thể LDL, tăng khả năng loại bỏ LDL-C khỏi máu và dẫn đến nồng độ cholesterol thấp. Đột biến mất chức năng ở gen ANGPTL3 làm tăng hoạt động của các enzyme phân giải lipid, dẫn đến giảm nồng độ LDL-C, triglyceride và HDL-C.
HBL là rối loạn di truyền đồng trội. Người mang một bản sao đột biến thường có triệu chứng nhẹ hoặc không có triệu chứng. Trong khi người mang hai bản sao đột biến có triệu chứng nặng hơn. Người mang đột biến dị hợp tử thường có LDL-C <80 mg/dL. Trong khi đồng hợp tử có mức LDL-C rất thấp (<20 mg/dL) hoặc không tổng hợp được apolipoprotein B, dẫn đến biến chứng nặng như gan nhiễm mỡ, thiếu hụt vitamin tan trong dầu.
Triệu chứng hạ betalipoprotein máu gia đình
Hạ betalipoprotein máu gia đình gây ra hàng loạt triệu chứng ảnh hưởng đến tiêu hóa, gan, thần kinh và mắt. Cụ thể là:
- Triệu chứng tiêu hóa bao gồm tiêu chảy mỡ, phân bạc màu và chướng bụng do cơ thể không hấp thu được chất béo và vitamin tan trong dầu (A, D, E, K). Ở trẻ em, tình trạng này có thể dẫn đến suy dinh dưỡng nặng, ảnh hưởng nghiêm trọng đến sự phát triển.
- Tổn thương gan cũng là một biểu hiện phổ biến của bệnh với các mức độ từ gan nhiễm mỡ, viêm gan cho đến xơ gan tiến triển. Tích tụ lipid trong tế bào gan làm tăng nguy cơ rối loạn chức năng gan và hình thành sỏi mật.
- Bên cạnh đó, người bệnh cũng có thể bị thiếu hụt vitamin. Vitamin E giảm dẫn đến thoái hóa võng mạc, thất điều tiểu não và yếu cơ. Thiếu vitamin A/K gây quáng gà và xuất huyết dưới da.
- Các triệu chứng thần kinh như mất điều hòa vận động, tê bì chân tay thường xuất hiện do tổn thương cột sống hoặc dây thần kinh ngoại biên.
- Ngoài ra, bệnh còn gây thoái hóa võng mạc nghiêm trọng, dẫn đến mù lòa nếu không được điều trị kịp thời.

Điều trị hạ betalipoprotein máu gia đình
Điều trị hạ betalipoprotein máu gia đình tập trung vào kiểm soát triệu chứng, ngăn ngừa biến chứng và bù đắp thiếu hụt dinh dưỡng.
Dinh dưỡng trị liệu
Người bị bệnh hạ betalipoprotein máu gia đình cần duy trì chế độ ăn cực ít chất béo (< 15% tổng năng lượng). Cần giảm tiêu thụ chất béo thông thường (dầu, mỡ động vật) để hạn chế tình trạng tiêu chảy phân mỡ và suy dinh dưỡng. Đồng thời, việc bổ sung dầu MCT (chất béo chuỗi trung bình) cũng là việc cần thiết. MCT được hấp thu trực tiếp vào tĩnh mạch cửa mà không cần chuyển hóa qua chylomicron, giúp cung cấp năng lượng và cải thiện hấp thu vitamin tan trong dầu.
Bổ sung vitamin
Vitamin E liều cao (100 - 300 IU/kg/ngày) được bổ sung giúp bảo vệ tế bào thần kinh và mô. Vitamin A (ngừa quáng gà), vitamin D (chống loãng xương), vitamin K (ngừa xuất huyết) dạng tan trong nước được bổ sung để khắc phục tình trạng kém hấp thu do thiếu lipoprotein.
Điều trị hạ betalipoprotein máu bằng thuốc
Acid nicotinic từng được nghiên cứu để tăng HDL, nhưng hiện không còn được khuyến cáo rộng rãi do hiệu quả lâm sàng hạn chế và tác dụng phụ. Ursodeoxycholic acid (UDCA) có tác dụng ngăn ngừa sỏi mật bằng cách giảm bão hòa cholesterol trong dịch mật và tăng lưu thông đường mật. Liều 10 - 15 mg/kg/ngày được khuyến cáo cho bệnh nhân có nguy cơ cao.

Mặc dù hạ betalipoprotein máu gia đình thường đặc trưng bởi mức cholesterol thấp, nhưng ở một số bệnh nhân có rối loạn chuyển hóa kèm theo, tình trạng mỡ máu cao vẫn có thể đồng thời hiện diện do nguyên nhân thứ phát. Trong những trường hợp này, người bệnh có thể cần được hỗ trợ điều trị bằng thuốc trị mỡ máu phù hợp, tùy theo tình trạng rối loạn chuyển hóa đi kèm.
Theo dõi biến chứng
Người bệnh cần đo mật độ xương 2 năm/lần để phát hiện sớm loãng xương và điều trị kịp thời. Kiểm tra thị lực hàng năm sẽ giúp tầm soát thoái hóa võng mạc. Ngoài ra, bệnh nhân cũng cần kiểm tra chức năng tiểu não định kỳ để phát hiện sớm thất điều hoặc rối loạn vận động. Xét nghiệm chức năng gan định kỳ để theo dõi diễn tiến gan nhiễm mỡ hoặc xơ hóa cũng là việc nên làm.
Phòng ngừa, kiểm soát hạ betalipoprotein máu gia đình
Phòng ngừa hạ betalipoprotein máu gia đình bắt đầu từ việc tư vấn di truyền, giúp đánh giá nguy cơ cho các gia đình có tiền sử bệnh. Ngoài ra, sàng lọc sơ sinh thông qua xét nghiệm APOB cho trẻ có cha/mẹ mang gen đột biến là một biện pháp quan trọng để phát hiện sớm và can thiệp kịp thời.
Mặc dù đây là một rối loạn di truyền không thể phòng tránh hoàn toàn, việc duy trì chế độ ăn uống cân bằng, giàu chất xơ và ít chất béo bão hòa có thể giúp điều chỉnh mức cholesterol trong cơ thể. Ngoài ra, việc tập thể dục đều đặn, giảm stress và kiểm soát cân nặng cũng đóng vai trò quan trọng trong việc giảm thiểu các biến chứng tim mạch do hạ betalipoprotein máu.
Hạ betalipoprotein máu gia đình không quá đáng ngại nếu được phát hiện sớm và can thiệp đúng hướng. Sự kết hợp giữa chế độ ăn ít chất béo, bổ sung vitamin liều cao cùng theo dõi đa chuyên khoa giúp cải thiện đáng kể chất lượng cuộc sống của bệnh nhân.
Xem thêm:
Có thể bạn quan tâm
Thông tin và sản phẩm gợi ý trong bài viết chỉ mang tính chất tham khảo, vui lòng liên hệ với Bác sĩ, Dược sĩ hoặc chuyên viên y tế để được tư vấn cụ thể. Xem thêm
Các bài viết liên quan
Top 7 ngũ cốc nguyên hạt giúp cải thiện cholesterol và cách ăn phù hợp
[Infographic] Cơ thể xử lý cholesterol như thế nào?
Xét nghiệm cholesterol là gì? Trường hợp nào nên xét nghiệm cholesterol?
Hướng dẫn cách đọc chỉ số cholesterol chính xác và dễ hiểu
Cholesterol và triglycerid khác nhau như thế nào?
HDL và LDL là gì? Phân biệt rõ ràng hai loại cholesterol trong máu
Thiếu HDL gia đình: Nguy cơ tiềm ẩn và những điều bạn cần biết!
Bệnh Tangier: Căn bệnh hiếm gặp nhưng nguy hiểm không ngờ!
Cholesterol có chức năng gì? Cách kiểm soát để phát huy vai trò của cholesterol
Nguyên nhân cholesterol cao là gì? Phương pháp phòng ngừa hiệu quả